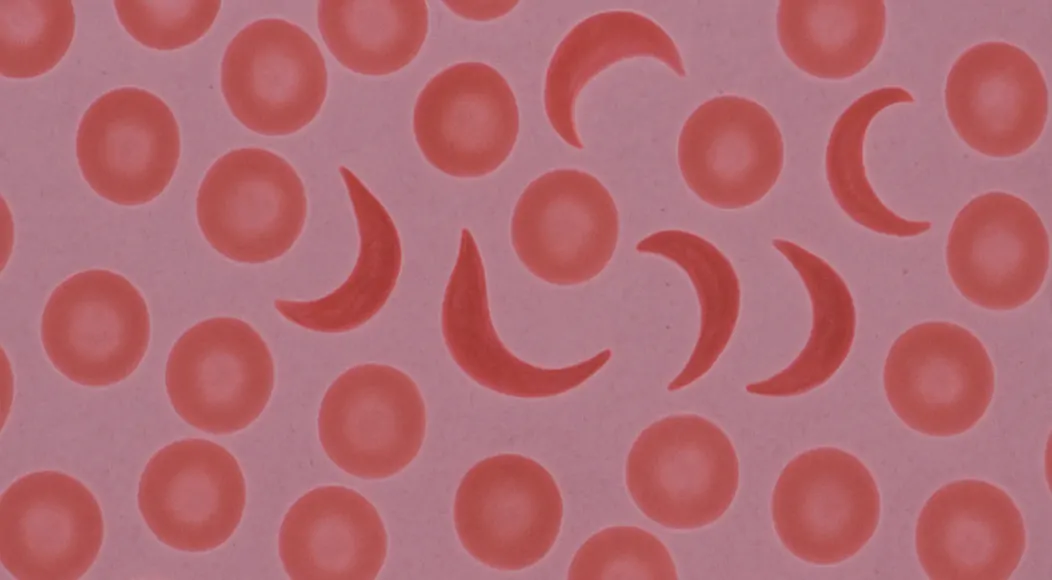
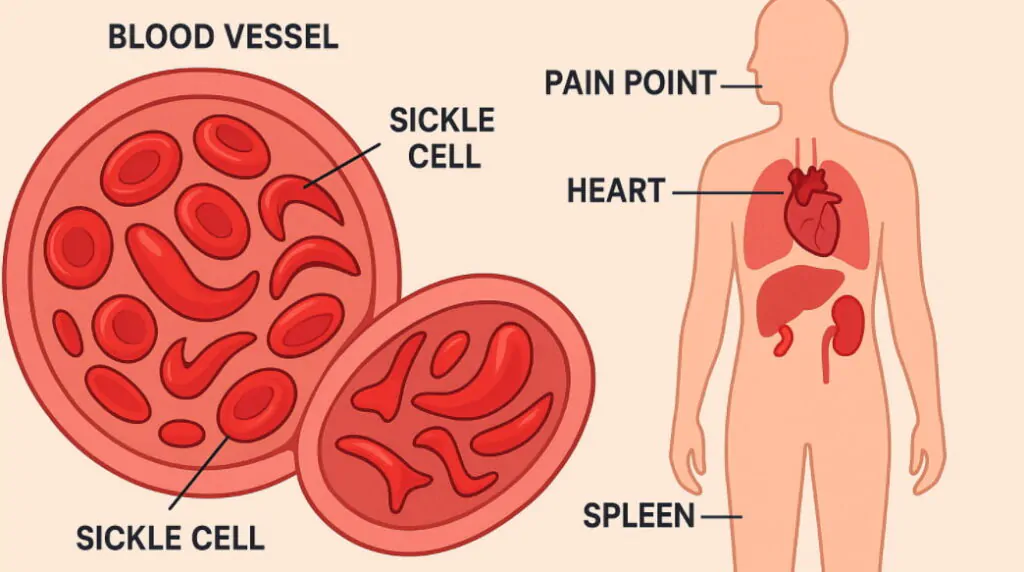
Sickle cell disease is a genetic blood disorder that affects the shape and function of red blood cells, causing them to become crescent or sickle shaped instead of their normal round form. What is sickle cell disease involves understanding how mutations in the HBB gene create abnormal hemoglobin, leading to cells that stick together and block blood flow throughout the body. Sickle cell anemia represents the most severe form of this inherited condition, affecting millions of people worldwide, particularly those of African, Mediterranean, and Middle Eastern descent. Sickle cell symptoms include severe pain episodes, fatigue, frequent infections, and organ damage that can significantly impact quality of life and require ongoing medical management. Genetic blood disorder inheritance occurs when both parents carry the sickle cell trait, creating a 25% chance of passing the full disease to their children. Recent advances in sickle cell treatment including gene therapy and CRISPR technology offer new hope for patients facing this challenging inherited blood condition that affects approximately 100,000 Americans and millions globally.
Important note: Educational content only. Not a substitute for professional medical advice diagnosis or treatment. Seek qualified care for personal decisions urgent symptoms or medication choices.
Key Clinical Features
- Chronic hemolytic anemia (pallor fatigue jaundice)
- Vaso occlusive pain episodes (often in bones chest abdomen)
- Dactylitis in young children
- Acute chest syndrome (respiratory symptoms with infiltrate)
- Splenic dysfunction increasing infection risk
- Delayed growth or puberty
- Stroke risk especially in childhood without screening
- Leg ulcers avascular necrosis retinopathy in older patients
Types and Variations of Sickle Cell Disease
Sickle cell anemia (HbSS) represents the most severe form, occurring when individuals inherit two sickle cell genes and experience the most frequent and intense symptoms.
Hemoglobin SC disease develops when one sickle cell gene combines with a hemoglobin C gene, typically causing milder symptoms but still requiring medical monitoring and treatment.
Sickle beta thalassemia results from inheriting one sickle cell gene and one beta thalassemia gene, creating varying severity levels depending on the specific thalassemia type involved.
| SCD Type | Genetic Combination | Severity Level | Common Symptoms |
|---|---|---|---|
| HbSS | Two sickle genes | Most severe | Frequent pain crises, organ damage |
| HbSC | Sickle + Hemoglobin C | Moderate | Less frequent crises, eye problems |
| Sickle Beta+ | Sickle + Mild thalassemia | Mild to moderate | Variable symptoms |
| Sickle Beta0 | Sickle + Severe thalassemia | Severe | Similar to HbSS |
Primary Symptoms and Complications
Pain crises occur when sickled cells block blood vessels, causing severe pain episodes that can last hours to days and require emergency medical treatment for proper management. Chronic anemia develops because sickled cells break down faster than normal red blood cells, leading to fatigue, weakness, and shortness of breath during daily activities. Organ damage affects the spleen, kidneys, liver, heart, and lungs over time as repeated blockages reduce oxygen delivery and cause progressive tissue deterioration.
Diagnosis and Screening Methods
Newborn screening programs in most countries now test all babies for sickle cell disease within the first few days of life, enabling early intervention and treatment planning.
Hemoglobin electrophoresis provides definitive diagnosis by separating different types of hemoglobin in blood samples, clearly identifying sickle hemoglobin and other variants present. Genetic counseling helps families understand inheritance risks, testing options, and reproductive choices when one or both parents carry sickle cell traits or have the disease.
Current Treatment Approaches
Hydroxyurea medication increases fetal hemoglobin production, reducing the frequency and severity of pain crises while protecting organs from damage over long-term use. Blood transfusions provide normal red blood cells to reduce complications, particularly important for preventing stroke and managing severe anemia in affected patients.
Pain management involves both preventive measures and acute treatment protocols using medications, hydration, and supportive care during painful episodes or crises.
Daily Self Care and Prevention Strategies
- Maintain hydration balanced with renal function.
- Prompt evaluation of fever (risk of sepsis).
- Adherence to medication and vaccine schedule.
- Early intervention for chest pain or breathing difficulty.
- Avoid extreme temperature changes and severe hypoxia (unpressurized altitude risk).
- Physical activity moderate with gradual conditioning.
Revolutionary Gene Therapy Advances
CRISPR gene editing technology recently received FDA approval through Casgevy, offering potential cures by editing patients’ own stem cells to produce normal hemoglobin.
Gene therapy options including bluebird bio’s Lyfgenia provide alternative approaches using modified viruses to introduce functional genes into patient bone marrow cells.
NHS approval for Casgevy in England marks a significant milestone, making cutting-edge gene therapy accessible to more patients through national healthcare systems.
Global Impact and Demographics
Population statistics show sickle cell disease affects approximately 100,000 Americans and over 20 million people worldwide, with highest prevalence in sub-Saharan Africa.
Geographic distribution correlates with historical malaria presence, as carrying one sickle cell gene provides protection against malaria, explaining the trait’s persistence.
Healthcare disparities affect access to specialized care and new treatments, particularly impacting underserved communities where sickle cell disease is most prevalent.
Living with Sickle Cell Disease
Daily management requires staying hydrated, avoiding extreme temperatures, managing stress, and maintaining regular medical appointments to prevent complications and monitor organ function.
Educational accommodations help students with sickle cell disease succeed academically while managing absences due to pain crises, fatigue, and medical appointments.
Career considerations may involve choosing jobs with flexible schedules, good health insurance, and understanding employers who accommodate medical needs and treatment schedules.
Prevention and Family Planning
- Carrier screening before pregnancy helps couples understand their risk of having children with sickle cell disease and make informed reproductive decisions.
- Prenatal testing options include chorionic villus sampling and amniocentesis to diagnose sickle cell disease in developing babies during pregnancy.
- Preimplantation genetic diagnosis allows couples using in vitro fertilization to select embryos without sickle cell disease, preventing transmission to future generations.
Research and Future Developments
Clinical trials continue investigating new medications, gene therapies, and treatment approaches aimed at reducing symptoms and potentially curing sickle cell disease.
Stem cell research explores using induced pluripotent stem cells and other cellular approaches to regenerate healthy blood-forming tissue in affected patients.
Global health initiatives work to improve access to basic care, newborn screening, and advanced treatments in regions where sickle cell disease is most common.
Support Resources and Organizations
Patient advocacy groups provide education, support, and resources for individuals and families affected by sickle cell disease while promoting awareness and research funding.
Healthcare teams typically include hematologists, pain specialists, social workers, and other professionals who collaborate to provide comprehensive care for complex needs.
Community programs offer peer support, educational workshops, and assistance with insurance navigation, transportation, and other practical challenges patients face.
Frequently Asked Questions
- Is sickle cell disease contagious? No, sickle cell disease is a genetic condition inherited from parents and cannot be caught from or transmitted to other people through contact.
- Can people with sickle cell disease have children? Yes, though genetic counseling is recommended to understand inheritance risks and discuss family planning options with healthcare providers.
- What triggers sickle cell pain crises? Common triggers include dehydration, infection, stress, extreme temperatures, high altitudes, and overexertion, though crises can occur without identifiable causes.
- How long do people with sickle cell disease typically live? Life expectancy has improved significantly with modern care, with many patients living into their 50s and beyond, especially with early diagnosis and treatment.
- Are there different severity levels? Yes, severity varies based on the specific genetic type, with sickle cell anemia being most severe and other variants causing milder symptoms.
- Can sickle cell disease be cured? Bone marrow transplant and newer gene therapies offer potential cures, though these treatments carry risks and aren’t suitable for all patients.
References and Source Links:
- Centers for Disease Control and Prevention https://www.cdc.gov
- World Health Organization https://www.who.int
- United States Food and Drug Administration https://www.fda.gov
- National Institutes of Health https://www.nih.gov
- National Health Service UK https://www.nhs.uk
- American Society of Hematology https://www.hematology.org
Article Summary
The expression what is sickle cell disease refers to a cluster of inherited hemoglobin disorders centered on hemoglobin S causing rigid sickled red cells chronic hemolysis and vaso occlusion. Hallmark features include anemia recurrent pain episodes infection vulnerability and progressive organ involvement. Modern care integrates newborn screening hydroxyurea transfusion strategies vaccination pain management and psychosocial support. Curative intent options now include matched donor transplantation and approved autologous gene therapy approaches like Casgevy and Lyfgenia with 2025 data showing quality of life improvement. Early multidisciplinary intervention equitable access and long term follow up of emerging therapies remain central public health priorities.